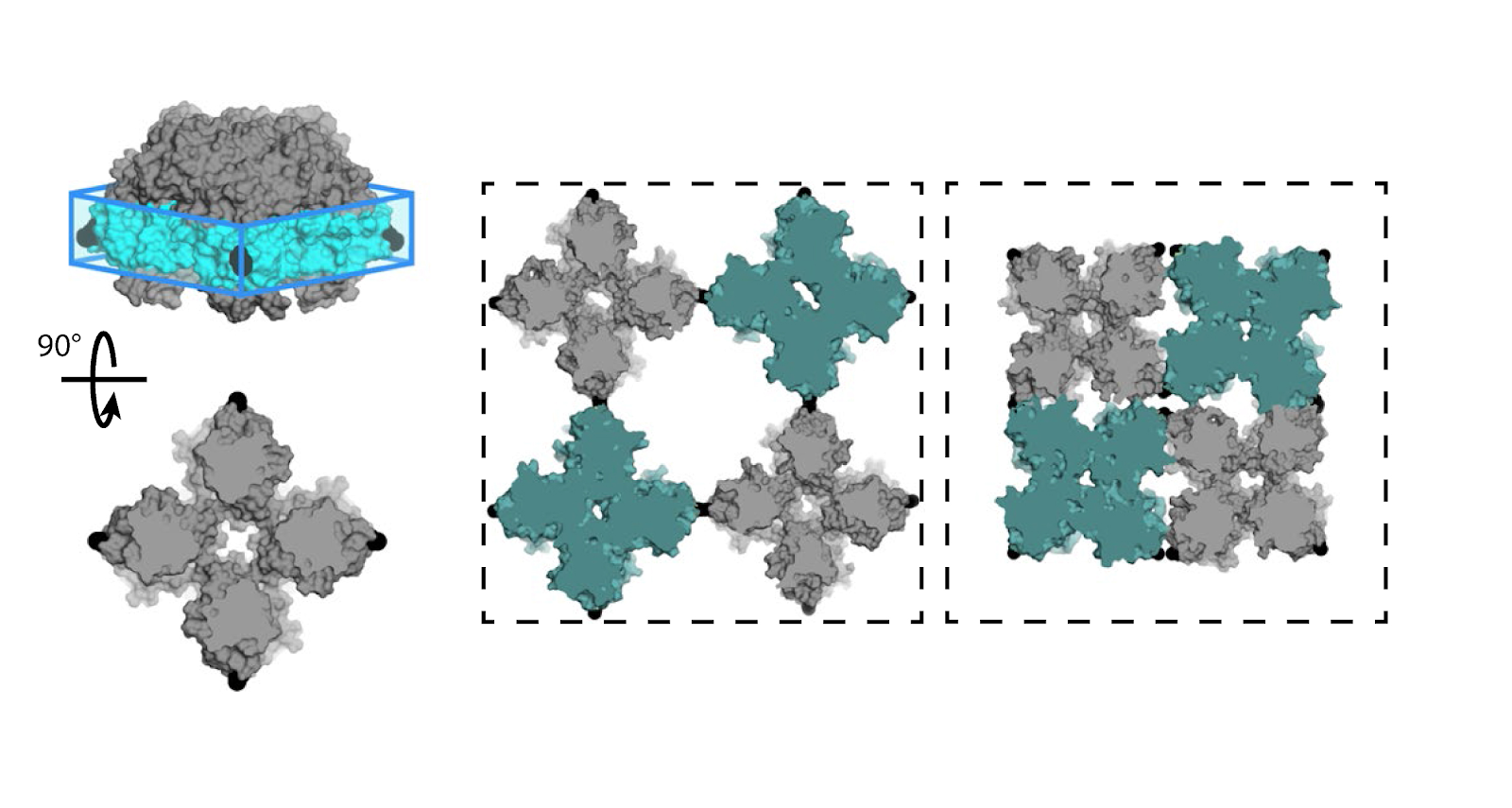
For the first time ever, chemists from the University of California, San Diego designed a two-dimensional protein crystal simulation that toggles between states of varying porosity and density. The research could help scientists create new materials for water purification, renewable energy, breakthroughs in medicine, drug development, and many more possible applications.
The work of combining experimental studies with computations executed on GPU-accelerated supercomputers is a first in biomolecular design, the researchers said.
“We did an extensive set of molecular dynamics simulations and experiments, which explained the basis of the unusual structural dynamics of these artificial proteins, based on which we were able to make rational decisions and alter the structural dynamics of the assembly,” said study co-author Akif Tezcan, a professor of chemistry and biochemistry at UCSD. “Our idea was to be able to build complex materials, like evolution has done, using proteins as building blocks,” Tezcan said.
To achieve the breakthrough, the team used over 130 NVIDIA Tesla GPUs on the Maverick supercomputer at the Texas Advanced Computing Center (TACC), and the GPU-accelerated Nanoscale Molecular Dynamics (NAMD) application, a production-quality molecular dynamics application designed for high-performance simulation of large biomolecular systems developed by University of Illinois at Urbana-Champaign (UIUC).
“The NAMD software that we use runs very well on GPUs. That allows us to speed up the calculations by orders of magnitudes,” explained study co-author Francesco Paesani, a professor of chemistry and biochemistry at UCSD. “Nowadays, we can afford calculations that ten years ago we couldn’t even dream about.”
The research was funded in part by the National Science Foundation and was recently published in the Nature Chemistry journal.
Read more>
Improving Biomolecular Design with GPU-Accelerated Supercomputers
May 18, 2018
Discuss (0)

Related resources
- GTC session: Accelerating Drug Discovery: Optimizing Dynamic GPU Workflows with CUDA Graphs, Mapped Memory, C++ Coroutines, and More
- GTC session: Enhancing Discovery in TechBio: Leveraging AI for Enzyme and Drug Discovery
- GTC session: Combining Quantum-Based Models With Machine Learning Accelerates Drug Discovery
- NGC Containers: CUDA
- Webinar: Bringing Drugs to Clinics Faster with NVIDIA Computing
- Webinar: Transforming Molecular Design